Table Of Content
- Underlying models of protein structure and function
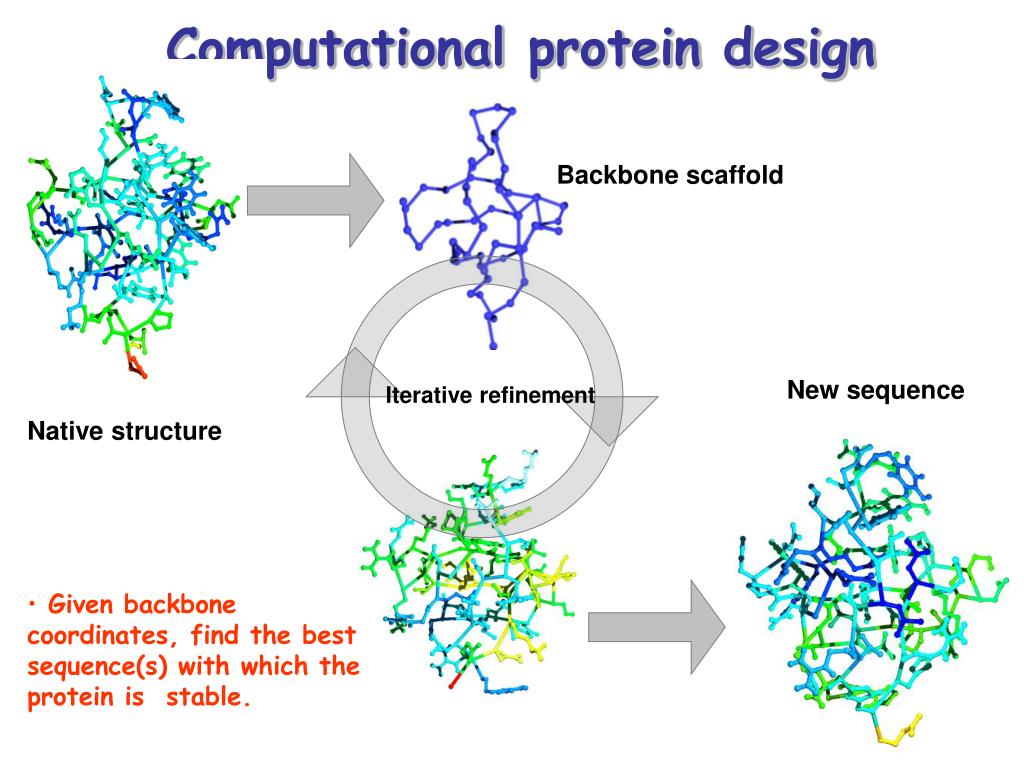
- SPDesign: protein sequence designer based on structural sequence profile using ultrafast shape recognition
- Protein Designs That Function In Vivo
- Enhancing the pharmaceutical properties of protein drugs by ancestral sequence reconstruction
- We create new proteins that solve modern challenges in medicine, technology, and sustainability.
- Protein design articles within Nature Biotechnology
Whereas some in silico success has been reported previously4, a general solution that can readily produce high-quality, orthogonally validated outputs remains elusive. Although RFdiffusion is unable to explicitly model bound small molecules at present (however, see our conclusions), the substrate can be implicitly modelled using an external potential to guide the generation of ‘pockets’ around the active site. As a demonstration, we scaffold a retroaldolase active site triad while implicitly modelling the reaction substrate (Extended Data Fig. 6e–h). Protein switches change their conformations when triggered by external signals, adding a potential extra layer of complexity over designing proteins that adopt multiple conformations.
Underlying models of protein structure and function
Tanja Kortemme, Professor of Bioengineering and Therapeutic Science, University of California San Francisco, has played a leading role in the field of protein design, with a focus on the invention of new approaches to engineer new biological functions at multiple scales. We develop protein design software and use it to create molecules that solve challenges in medicine, technology, and sustainability. By iterating between computation and laboratory experiments, we continually improve our protein design methods. B.L.T. and J.Y., with assistance from V.D.B. and E.M., extended diffusion to residue orientations.
SPDesign: protein sequence designer based on structural sequence profile using ultrafast shape recognition
We release OpenCRISPR-1 publicly to facilitate broad, ethical usage across research and commercial applications. Proteins perform functions by placing atoms with certain physicochemical properties at specific positions in the three-dimensional space. Initial work on the functional protein design directly borrowed from native functional site “motifs” (three-dimensional arrangements of functional groups in an existing active site) (136).
Protein Designs That Function In Vivo
After generation of protein backbones, the second step in a typical de novo protein design protocol is selection of amino acid side-chain types and conformations to stabilize the backbone conformation and to adopt specific three-dimensional active site geometries optimized for function. Early de novo design studies used amino acids that favor specific secondary structure types (85) or binary polar/hydrophobic patterns (86) to define protein structures. Because side-chain conformations are clustered as rotamers (87, 88), the side-chain design can be formulated as a discrete optimization problem (89), that is, find a combination of rotamers that minimize the energy of a structure. The complexity of the problem grows exponentially with the increase of the number of residues. Small-scale side-chain design problems can be solved deterministically by the dead-end elimination algorithm (90), but many de novo protein side-chain optimization problems are too large to be solved deterministically. Instead, amino acid sequences and side-chain conformations are often optimized using Monte Carlo methods (91, 92), which do not guarantee to find the global minimum, but the solutions are often sufficiently accurate for applications.
Targeting MYC with modular synthetic transcriptional repressors derived from bHLH DNA-binding domains
Hydrogen bonds play an important role in the specificity of protein–ligand and protein–protein interactions. The formation of a hydrogen bond only allows narrow ranges of distances and orientations between the donor and acceptor groups (38). Almost all hydrogen bond donor or acceptor groups in a protein must form hydrogen bonds within the protein or with solvent molecules to avoid large energetic penalties of unsatisfied hydrogen bonds (99). The HBNet method addresses the challenges for the hydrogen bond design by systematically searching for possible hydrogen-bond networks (100) (Fig. 3D).
Even small proteins (100 residues or less) have hundreds of backbone degrees of freedom, making it impossible to sample the backbone structure space by brute force. Moreover, because folded proteins need to have well-packed cores and satisfied hydrogen bonds, only a small fraction of the backbone structure space can stably exist, that is, is “designable” (47, 48). The design of a functional de novo protein, for example, a binder (middle, magenta) to a target protein (middle, gray), requires sampling of the backbone structure space to find a backbone compatible with the function, sequence optimization to stabilize the backbone, and designing the functional site interactions. A scoring function is necessary to select designs with desired properties, typically by identifying low-energy sequence–structure combinations. Protein function is heavily dependent on protein structure, and rational protein design uses this relationship to design function by designing proteins that have a target structure or fold. Thus, by definition, in rational protein design the target structure or ensemble of structures must be known beforehand.
Enhancing the pharmaceutical properties of protein drugs by ancestral sequence reconstruction
Proteins with controllable curvatures can be designed by combinations of modular leucine-rich-repeat units (65) (Fig. 2A). The structure extension with native-substructure graphs (SEWING) method (36) combines continuous or discontinuous helical building blocks from existing proteins (Fig. 2A). Substructures that share high similarity in local regions are overlapped and combined. Notably, previous applications of Crick’s parameterization to the design were restricted to the coiled-coil topology, while SEWING allows the exploration of more diverse helical topologies.
If researchers used AI in this process at all in recent years, it was primarily to improve existing molecules. Tess van Stekelenburg, an investor at Hummingbird Ventures, notes that Basecamp — one of the companies funded by the firm — captures all manner of environmental and biochemical context for the proteins it identifies. The resulting ‘metadata’ accompanying each protein sequence can help guide the engineering of proteins that express and function optimally in particular conditions. “It gives you a lot more ability to constrain for things like pH, temperature or pressure, if that’s what you’re planning to look at,” she says. The supplementary information file is a single PDF that contains text, figures and tables that aim to help the reader understand the theoretical underpinnings of RFdiffusion, its implementation and its application to the design challenges posed in the paper. Efforts to design new proteins were first undertaken with the intent to increase our knowledge of structure and activity but also with the promise of creating new practical protein tools.
Protein design articles within Nature Biotechnology

The ability to explore such geometric variation within fold families is critical for design of new protein functions that require precise three-dimensional conformations of active sites. The recently developed loop-helix-loop unit combinatorial sampling method systematically samples loop-helix-loop geometries in arbitrary protein folds by near exhaustive testing of combinations of short loops (32) (Fig. 2D). The generated protein geometries had similar distributions to those observed in native structures in the PDB but also included thousands of new structures. Using a different approach to geometric variation, an enumerative algorithm was developed to sample diverse pocket structures of nuclear transport factor 2 fold proteins (28).
At the same time, however, these energy functions must consider the computational challenges behind protein design. One of the most challenging requirements for successful design is an energy function that is both accurate and simple for computational calculations. Thus, an essential parameter of any design process is the amount of flexibility allowed for both the side-chains and the backbone.
They have already succeeded in creating proteins with novel catalytic functions for use in agriculture, materials and food science. These projects often begin with a relatively well-established core reaction that is catalyzed in nature. But to adapt these reactions to work with a different substrate, “you need to remodel the active site dramatically,” says Zanghellini. Some of the company’s projects include a plant enzyme that can break down a widely used herbicide, as well as enzymes that can convert relatively low-value plant byproducts into useful natural sweeteners.







No comments:
Post a Comment